FastQC
Perform QC from your reads prior to other NGS processing
![]() suggests : cutadapt, seq_crumbs, PrinSeq, FastX_toolkit
suggests : cutadapt, seq_crumbs, PrinSeq, FastX_toolkit
FastQC([1])) aims to provide a simple way to do some quality control checks on raw sequence data coming from high throughput sequencing pipelines. It provides a modular set of analyses which you can use to give a quick impression of whether your data has any problems of which you should be aware before doing any further analysis (watch the tutorial video here[2]).
FastQC is a very popular standalone Java GUI application running on all platforms.
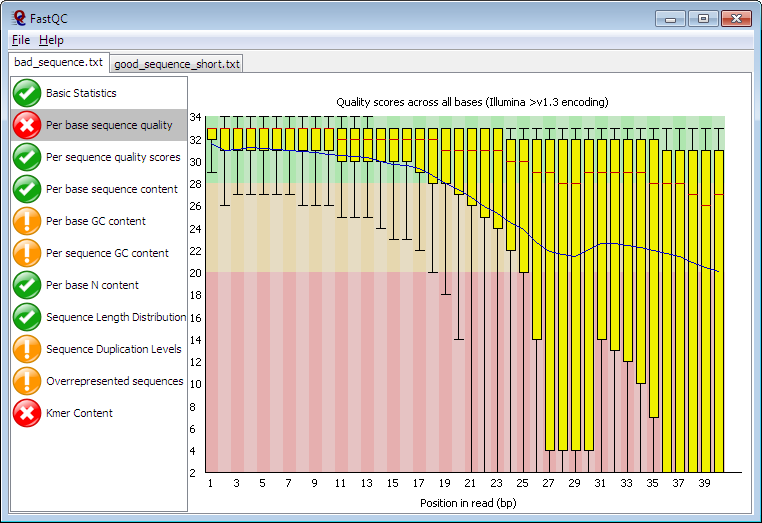
![]() FastQC also runs as a command line to submit runs in batch and store the html reports as zip files for later review (see: Perform basic read QC at command line prior to mapping)
FastQC also runs as a command line to submit runs in batch and store the html reports as zip files for later review (see: Perform basic read QC at command line prior to mapping)
The main functions of FastQC are:
- Import of data from BAM, SAM or FastQ files (any variant)
- Providing a quick overview to tell you in which areas there may be problems
- Summary graphs and tables to quickly assess your data
- Export of results to an HTML based permanent report
- Offline operation to allow automated generation of reports without running the interactive application
References:
- ↑ http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ↑ http://www.youtube.com/watch?v=bz93ReOv87Y